



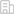

家蚕基因组生物学国家重点实验室主任、国家蚕桑产业技术体系首席科学家代方银教授团队完成家蚕大规模种质资源基因组解析(“千蚕基因组”),绘就家蚕超级泛基因组。该研究在世界上率先实现家蚕基因库数字化,创建“数字家蚕”,对于深化功能基因组研究和推进家蚕模式化,开启家蚕设计育种,赋能“改造家蚕、多元利用”等,将产生深远的影响。
家蚕是重要的经济昆虫和新兴模式生物,但之前仅有单一参考基因组及部分重测序,尚不足以支撑基因组变异和优良基因的深度挖掘,尤其对于分子育种具有显著局限。超级泛基因组是一个物种所有基因组信息的总和,对于深化功能基因组研究、种质创新等意义重大。
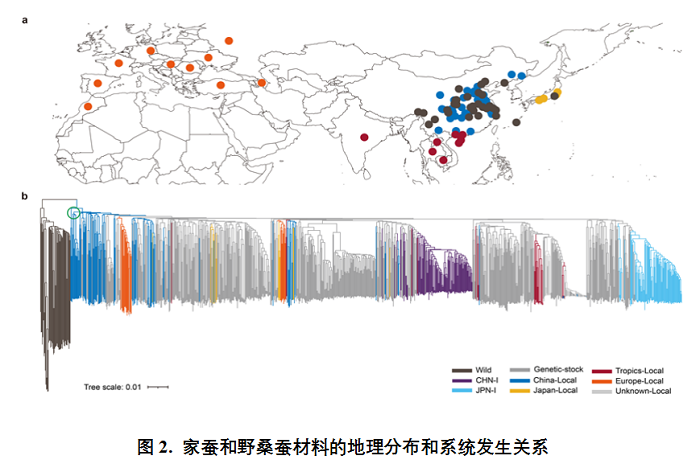
研究团队对1078份蚕种质资源(205份地方种,194份改良种,632份遗传材料,47份野桑蚕)进行了深度二代(短读长)测序,对其中545份代表性资源进行了三代(长读长)测序,产生55.57T基因组数据,组装了545个蚕的高质量基因组,对100个基因组进行了基因注释,鉴定到4300余万个SNP、930余万个Indel、340余万个结构变异(SV)和7308个新基因(家族),绘制了一个高精度家蚕泛基因组图谱。该超级泛基因组囊括了目前最全面的家蚕和野桑蚕基因组信息,是迄今全球动植物领域最大的长读长泛基因组。同时,对蚕的各种遗传变异、群体结构、人工选择和生态适应性及经济性状开展了深入的研究,取得丰硕的创新结果。研究论文“High-resolution silkworm pan-genome provides genetic insights into artificial selection and ecological adaptation”于2022年9月24日在国际名刊Nature Communications在线发表。


家蚕起源于我国,由中国古野桑蚕驯化而来,具有5000多年的驯养历史,但其驯化起源地长期是悬而未决的问题,特别是缺乏有力的生物学证据。本研究的材料代表了来自全球各主要养蚕地区的最丰富的遗传多样性,研究发现黄河中下游地区的地方种分布在进化树上家蚕分支的基部,表明家蚕起源于黄河中下游地区。现有的考古证据,包括1926年在山西夏县西阴村出土的半颗蚕茧,2019年在该县师村出土的石雕蚕蛹等都为此结论提供了重要的佐证。
蚕的传统育种历史悠久且成就卓著,但自上世纪90年代以来已遭遇瓶颈。系统解析驯化与改良选择作用的遗传基础,对于突破家蚕育种瓶颈极为重要。研究团队鉴定到468个驯化相关基因和198个改良相关基因,其中新鉴定分别为264和185个。这些基因将是家蚕分子改良的重要候选靶标。同时,发现中国实用种和日本实用种只共享不到3%的改良作用位点,这不仅揭示了中国和日本相对独立的家蚕育种历史,而且解释了中日两个系统间产生强杂交优势的遗传基础奥秘。这一结果对家蚕的现代育种具有重要启示。

长期以来家蚕育种选择的主要目标经济性状是茧丝相关性状,如茧丝产量和品质性状。然而,迄今为止对这类属于数量遗传性状的控制基因和位点知之甚少。泛基因组可谓是连接表型特别是复杂性状和序列之间“最近的桥”。在本研究中的一个案例,从选择信号和结构变异切入,揭示了与细胞周期相关的转录因子BmE2F1调控家蚕茧丝产量。通过CRISPR-cas9敲除BmE2F1后,蚕的丝腺细胞数减少了7.68%,产丝量减少22%;在丝腺中过表达BmE2F1后,丝腺细胞数增加了23%,产丝量增加16%。另一方面,茧丝纤度是蚕丝的重要品质性状,细纤度蚕丝具有独特应用和更高的经济价值,但之前对茧丝纤度的分子遗传基础毫无所知。本研究中的另一个案例,通过分析细纤度品种基因组中存在的稀有变异,鉴定到控制茧丝纤度的基因BmChitβ-GlcNAcase,该基因在细纤度品种中表达量显著提高,敲除该基因后家蚕的茧丝纤度变粗,表明该基因在茧丝纤度的决定中起关键作用。

滞育是昆虫中常见的一种生态适应性性状,它能确保昆虫顺利躲过不利于生存的环境条件。虽然滞育激素早在1957年于家蚕中首次被发现(Nature,1957),但至今关于胚胎滞育基因的信息却很少。本研究中,基于家蚕“着色非滞卵”突变体(pnd)和基因组结构变异分析,并通过基因编辑进行功能验证,揭示了BmTret1-like基因是重要的胚后滞育决定因子。这是昆虫中首次鉴定出胚后滞育决定基因。
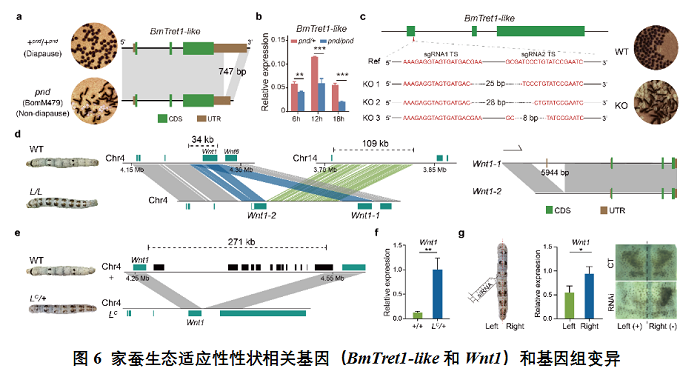
警戒色是昆虫另一种重要的生态适应性性状。家蚕中的褐圆斑(L)和菱纹茶斑(LC)突变体幼虫具有相似的成对分布的特殊斑纹,这种模式的斑纹在昆虫中通常用作恐吓天敌的警戒色。此前有研究推测L表型可能是由4号染色体上Wnt1基因上游19 kb内的小片段变异(SNPs and/or InDels)引起的(Nat.Commun,2013)。在本研究中,基于新组装的大量品系的高质量基因组,分析发现L突变体中特异存在两个大片段结构变异,其中,一个34 kb基因组重复包含了一个额外的Wnt1拷贝,在两个Wnt1拷贝之间还插入了一个来源于14号染色体的109 kb大片段。对于等位突变LC,则在Wnt1的3’侧翼区域发现了一个271 kb的特异性大片段缺失。这些在Wnt1基因邻近区域检测到的大片段的插入、缺失、重复等复杂变异,是之前的研究未能识别到的,显示出泛基因组在复杂变异解析中具有独特优势,特别是能提供更全面和准确的基因组信息。
论文评审专家认为:“该研究揭示了蚕完整泛基因组,会让家蚕研究人员睁大眼睛、高兴得跳起来。”“这项研究的优势在于它的全面采样,详细描述的泛基因组特性,以及对家蚕重要生物学性状的系统解析及功能研究;建立如此广泛的数据集,并结合各种实验来研究蚕的驯化问题是值得称赞的。”“这些令人印象深刻的数据集必将促进昆虫生物学和进化的研究,也必将提高家蚕育种的水平。”“论文详细描述了泛基因组的分析过程,对非家蚕研究人员也有很好的参考价值。”
西南大学家蚕基因组生物学国家重点实验室童晓玲教授、韩民锦副教授、陆昆鹏博士后,深圳华大基因科技有限公司高级工程师太帅帅、西南大学博士研究生梁书博和中国科学院遗传与发育研究所博士后刘羽诚为论文共同第一作者,西南大学家蚕基因组生物学国家重点实验室代方银教授为论文最后通讯作者,向仲怀院士、鲁成教授、童晓玲教授,以及西北工业大学王文教授、中国科学研究院遗传与发育研究所田志喜研究员为论文共同通讯作者。主要合作单位还有重庆市蚕业科学研究院、江苏科技大学、沈阳农业大学、安康学院、广东省农业科学研究院蚕业与农产品加工研究所、新加坡国立大学、法国斯特拉斯堡大学。国内多家有关蚕业科研院所为本研究的实验材料优化构成等提供了支持,已在论文中一并致谢。本研究受到国家自然科学基金重点项目(31830094、U20A2058)、国家现代农业产业技术体系项目(CARS-18-ZJ0102)、重庆市自然科学基金创新群体项目(cstc2021jcyj-cxtt0005)等资助。