



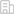

- 223 高校采购信息
- 1313 科技成果项目
- 10 创新创业项目
- 0 高校项目需求
多靶点协同抗癌候选新药Thiotert项目


Thiotert(研究代号命名YLS004)是浙江大学药学院余露山教授研究开发的抗肿瘤新药,研究表明,其同时具有硫氧还蛋白-1(Trx-1)、端粒酶和抗代谢抗肿瘤机制等多靶点协同增效作用。体内外研究结果表明,Thiotert对结直肠癌、黑色素瘤和白血病等具有显著的疗效,与多种临床一线用药相比在药效和毒性方面表现出明显的优势。该新药已获得专利授权(ZL201811003442.6),并获得国内同行专家的高度评价,获得浙江省重点研发计划专项(2020C03045)和中国药学会-以岭生物医药创新基金(2020年)的资助。目前,该候选新药正在洽谈融资拟进行临床前申报研究。

以下是Thiotert针对硫氧还蛋白-1和端粒酶两个靶点的相关材料,其对白血病的研究还在进行中,结果未整理到该文件。
(一)国内外研究现状和发展趋势。
据2019年1月我国最新的癌症统计数据显示,2015年(统计数据晚3年)我国恶性肿瘤发病约392.9万人,死亡233.8万人,恶性肿瘤死亡占居民全部死因的23.9%。而2018年全球范围内有1810万癌症新发病例和960万癌症死亡病例。由于肿瘤的高度异质性,一些抗肿瘤药物只能对某种或是某类肿瘤有效,而且随着重复给药的进行又非常容易耐药。同时,许多肿瘤特别是耐药性肿瘤常需要两种或多种化疗药物联合用药后才能被有效抑制。这说明,这些肿瘤需要从多个靶点同时给药才能起到良好的抑制疗效。因此,针对双/多靶点协同的抗肿瘤药物设计是抗癌新药研发的热点。
- 1. 硫氧还蛋白-1及抗癌药物研究
硫氧还蛋白-1(Thioredoxin-1,Trx-1)是一种促进肿瘤生长,抑制细胞凋亡,能上调低氧诱导因子HNF-1α和血管内皮生长因子VEGF的低分子量的细胞氧化还原蛋白。在乳腺癌、宫颈癌、肺癌、肝癌,结直肠癌、胃癌和黑色素瘤等肿瘤病人的血液和癌组织中Trx-1的浓度都较正常人体中显著升高。Trx-1在肿瘤细胞对化疗药物产生耐药性方面发挥着重要的作用,如Trx-1含量越高的成人T细胞白血病细胞对阿霉素的敏感性越低,多烯紫杉醇敏感的MCF-7乳腺癌细胞转染Trx-1基因后对多烯紫杉醇耐药,而敲低膀胱癌和前列腺癌细胞中Trx-1后显著增加它们对丝裂霉素C的敏感性等。因此,Trx-1靶点是抗肿瘤药物设计的热门靶点。
虽然目前临床上还没有针对该靶点的上市新药,但是已有多个候选新药正在临床研究中。比如,由北京大学研发的乙烷硒啉正在进行临床II期研究(登记号CTR20140581),其是通过抑制硫氧还蛋白还原酶进而抑制Trx-1水平,临床定位胃癌、肺癌和结肠癌。莫特沙芬钆也是一种针对该靶点的候选新药,目前正在进行临床III期,对脑癌具有显著的疗效。金诺芬是一种广泛用于治疗类风湿性关节炎的药物,但研究发现其可通过Trx-1靶点对多种肿瘤发挥显著疗效,目前美国同步开展了4个临床试验对金诺芬进行抗肿瘤药效的二次开发(临床登记号NCT01419691,NCT01737502,NCT017477798和NCT03456700)。
PX-12(1-甲基丙基-2-咪唑基二硫醚)也是一种能抑制Trx-1的新药分子,其分子中的二硫键是该药物分子与Trx-1发生反应的关键活性区域。-Cys-Gly-Pro-Cys-是Trx-1的保守催化位点,其主要通过Cys32和Cys35两个半胱氨酸残基发挥功能。Trx-1能将这两个半胱氨酸残基氧化成二硫醚,进而导致Trx-1失活,作用机制如下图所示。

临床前研究数据表明,PX-12对乳腺癌细胞MCF-7和胃肠道癌如结直肠癌细胞HT-29具有显著的抑制作用。I期临床试验数据显示,PX-12能剂量依赖性有效降低多种肿瘤病人循环血液中Trx-1和VEGF水平,并与肿瘤病人的生存期具有明显的相关性。按300 mg/m2/天的给药剂量连续三天静脉注射给与肿瘤病人志愿者,病人仅出现了1-2级的副作用,没有出现3和4级的副作用,报道的副作用为可逆性缺氧和肺炎。以上结果提示,PX-12不但具有显著的抑瘤效果,同时临床副作用很小,显示良好的抗肿瘤开发前景。
- 2. 端粒酶及抗癌药物研究
端粒位于真核线性染色体末端,其作用是保持染色体的完整性和控制细胞分裂周期。由于“末端复制问题”、氧化损伤和其他复制相关的末端处理事件,端粒在正常体细胞每轮DNA复制中会逐渐缩短。端粒酶通过在细胞中的线性染色体末端添加六聚体端粒DNA(TTAGGG)重复来减少端粒缩短。因而,在端粒酶阴性的细胞中,因为端粒缩短,细胞会进行复制性衰老,但在端粒酶阳性的细胞中,细胞会永生化,有可能发展为癌症。除了生殖细胞,干细胞,激活的淋巴细胞外,大多数正常的人体细胞不具有端粒酶活性,但在约85%至90%的原发性人类癌症中几乎普遍检测到它。因此,通过抑制端粒酶活性就能抑制肿瘤的无限繁殖,许多的研究致力于寻找有效的端粒酶抑制剂。
国内外针对端粒酶抑制剂的研究主要包括4个方面:抑制端粒酶活性,抑制端粒酶的疫苗,抑制端粒酶的病毒,以及破坏端粒完整性。目前,虽然还没有针对端粒酶的上市药物,但是已有多个候选药物正在进行临床前研究和临床各期试验,分别是GRN163L,GV1001,GRNVAC1,G-quadruplex ligands和TERT promoter-driven oncolytic virus等。然而,这些候选药物都是大分子化合物。
学术期刊Cancer Discovery(IF 26.37)报道了一种有效的小分子端粒酶抑制剂6-硫代-2'-脱氧鸟苷(6-thio-2′-deoxyguanosine,6-thio-dG),它可被端粒酶识别,并被整合到从头合成的端粒中,导致端粒功能障碍,抑制肿瘤增殖(Cancer Discov, 2015, 5: 82-95)。这是至今为止报道的唯一一个小分子端粒酶抑制剂。据最新的Clarivate Analytics新药研发进度网站查询,预计6-thio-dG将在2019年第3季度进入临床I期试验阶段。已有的临床前研究结果表明,6-thio-dG对I/O-、BRAF-、EGFR靶向治疗难治性肿瘤具有广泛的抑制作用,尤其对结直肠癌、肺癌、恶性黑色素瘤具有明显的疗效。特别是,在靶向分子疗法中6-thio-dG能透过小鼠血脑屏障,选择性杀死不可治愈类型儿童脑癌,包括固有桥脑胶质瘤,高级胶质瘤和高危髓母细胞瘤,表现出非常好的效果。
综上所述, Trx-1和端粒酶在大多数肿瘤中的表达都显著提高,因此针对这两个靶点的抗癌药物可能具有广谱抗癌作用。由于肿瘤的异质性,多靶点联合用药对大部分肿瘤特别是耐药性肿瘤常表现明显的效果,是临床化疗的主流方案。为此,我们提出了这样一个假设,能否将这两种治疗靶点设计在同一个化合物上,让它同时具有Trx-1和端粒酶抑制作用,从而达到协同抑制肿瘤的效果呢?依据这一个思路,我们前期对PX-12和6-thio-dG母核进行了结构修饰,获得的新化合物对结直肠癌和黑色素瘤具有显著的抑制作用,且经评估成药性好,具有良好的开发前景。
(二)项目主要研究开发内容、关键技术及主要创新点。
1.研究成果和研发内容
1.1.已有研究成果
按照上述研究思路,我们设计了4个目标化合物,结构如下:

对这些化合物初步的性状研究发现,它们为乳白色粉末,对光较为稳定。具有良好的溶解度,其中YLS004在水溶液中的溶解度远大于10 mg/mL,适合做成粉针剂。
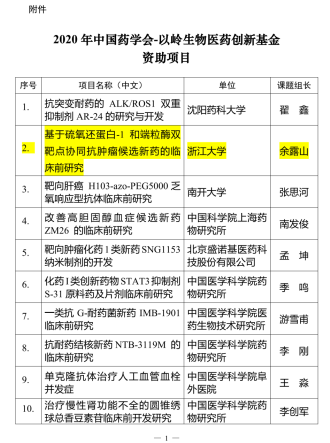
体外机制实验结果表明,这4个化合物都保留了较好的Trx-1活性抑制作用,其中YLS004的抑制作用与PX-12相当,结果见表1。同时,YLS002和YLS004保留了与6-Thio-dG的端粒酶逆转录酶(TERT)抑制作用以及对DNA和端粒的损伤作用,作用略强于对照化合物,见图1。以上结果表明,我们设计的化合物同时保留了Trx-1活性抑制和端粒酶抑制作用,与预期结果一致。
表1. 2-脱氧鸟苷类衍生物的Trx-1活性抑制作用
|
化合物 |
YLS001 |
YLS002 |
YLS003 |
YLS004 |
6-thio-dG |
PX-12 |
克拉屈滨 |
|
IC50(μM) |
31.71 |
9.78 |
47.73 |
4.26 |
>100 |
2.50 |
>100 |
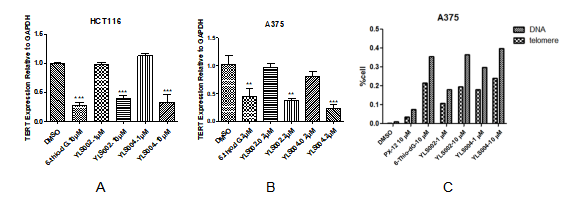
图1. YLS002和YLS004对端粒酶逆转录酶(TERT)的抑制以及对DNA和端粒的损伤作用。图A和B为在人结肠癌细胞HCT116和黑色素瘤细胞A375上YLS002和YLS004对TERT的抑制作用;图C为对DNA和端粒的损伤作用。
以6-thio-dG、PX-12和上市结构类似抗癌药物克拉屈滨(Cladribine)作为阳性对照药,受试化合物对人结肠癌细胞HCT116、SW620、HT29和人恶性黑色素瘤细胞A375都具有显著的抑制作用,特别是YLS004其抑制作用都优于阳性对照药物,而对正常结肠上皮细胞未表现抑制作用,结果见表2。而且,YLS004对HCT116和A375细胞的IC50值明显低于6-Thio-dG+PX-12合用组,见图2。该结果表明,YLS004体现了Trx-1和端粒酶抑制的显著抑瘤协同作用。
表2. 2-脱氧鸟苷类衍生物的肿瘤细胞生长抑制作用
|
化合物 IC50(μM) |
YLS001 |
YLS002 |
YLS003 |
YLS004 |
6-thio-dG |
PX-12 |
克拉屈滨 |
|
NCM460 正常结肠上皮细胞 |
>1000 |
>1000 |
>1000 |
>1000 |
>1000 |
>1000 |
>1000 |
|
HCT116 人结肠癌细胞 |
1.27 |
2.66 |
21.82 |
1.09 |
17.50 |
34.54 |
28.45 |
|
SW620 人结肠癌细胞 |
0.21 |
1.71 |
5.43 |
0.11 |
>100 |
1.68 |
>100 |
|
HT29 人结肠癌细胞 |
4.55 |
3.02 |
7.37 |
4.24 |
3.04 |
3.37 |
9.44 |
|
A375 人恶性黑素瘤细胞 |
0.60 |
0.52 |
3.06 |
0.37 |
0.40 |
2.98 |
/ |
|
BGC 胃腺癌细胞 |
6.09 |
/ |
/ |
11.70 |
>100 |
/ |
/ |
|
K562 白血病细胞 |
9.885 |
10.134 |
28.732 |
15.732 |
23.342 |
21.657 |
7.863 |
|
HepG2 肝癌细胞 |
10~50 |
<10 |
10~50 |
<10 |
<10 |
10~50 |
/ |
|
A549 肺癌细胞 |
30.8 |
13.52 |
35.79 |
31.12 |
33.59 |
7.43 |
/ |
|
MCF7 乳腺癌细胞 |
/ |
15.56 |
/ |
41.31 |
>100 |
>100 |
>100 |
|
Bcap37 乳腺癌细胞 |
18.24 |
21.04 |
35.12 |
22.77 |
>100 |
>100 |
>100 |
注:/,表示未进行该组实验。
图2. YLS004与6-Thio-dG+PX-12合用组分别对HCT116和A375细胞的抑制作用。
体内HCT116裸鼠荷瘤实验表明,当给药剂量以相同摩尔数给药时,与其他3个受试化合物相比,YLS004具有更好的肿瘤抑制作用,而且显著优于6-thio-dG和PX-12,见图3。血液生化指标显示,该剂量下各组都未见明显的毒性(见工作基础4,表3)。以上体内外的研究结果表明,YLS004的分子设计保留了Trx-1和端粒酶抑制作用,药效上显示了显著的双靶点协同抑瘤作用。
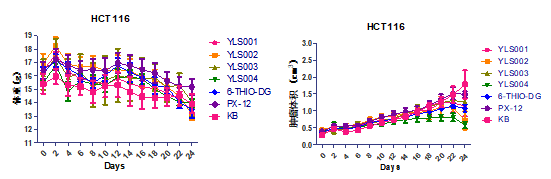
图3. HCT116细胞荷瘤鼠模型给药过程中各组裸鼠体重和肿瘤体积的变化
此外,我们采用人恶性黑色素瘤细胞A375荷瘤鼠模型进一步研究了YLS004对人恶性黑色素瘤的抑制作用。该实验以人恶性黑色素瘤的临床一线用药达卡巴嗪(Dacarbazine,DITC)为阳性对照药物(其给药剂量为20.40 mg/kg,相当于10 mg/kg YLS004给药剂量摩尔数的4倍),同时设置了6-thio-dG + PX-12联合用药组(给药剂量与10 mg/kg YLS004等摩尔剂量)。结果发现,按摩尔数计算,YLS004给药剂量为阳性对照药DITC给药剂量的四分之一时,其抑瘤作用都显著高于DITC,而且其抑瘤作用也显著高于同摩尔给药剂量下6-thio-dG和PX-12联合用药组(见图4)。该结果表明,YLS004的抑制人恶性黑色素瘤活性要显著高于临床一线用药DITC,而且从大鼠体重来看,虽然给药初期YLS004组体重下降较快,但是给药6天后体重开始逐渐升高,到给药13天后,体重明显超过DITC组、6-thio-dG和PX-12联合用药组,表明连续给药后机体能快速逆转该毒性。此外,从与6-thio-dG和PX-12联合用药组的抑瘤作用和体重变化相比,YLS004比联合用药组抑瘤作用显著增加,而且毒性更低。在ICR小鼠上以25 mg/kg/天的给药剂量连续给药5天后,仅在造血系统和脾脏发现了轻微的毒性(见工作基础7)。以上结果说明,YLS004的对人恶性黑色素瘤的抑瘤作用和毒性都显著优于6-thio-dG+PX-12两种药物的同摩尔剂量物理组合以及临床一线用药DITC(4倍摩尔剂量),同样体现了双靶点的显著协同抑瘤作用。
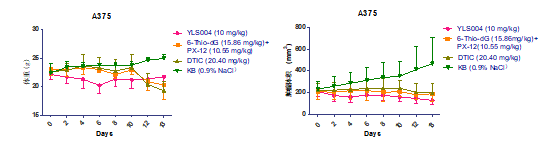
图4. YLS004对A375细胞荷瘤鼠模型给药过程中各组裸鼠体重和肿瘤体积的变化
Trx-1和端粒酶在多种肿瘤细胞中都高表达,它们对肿瘤的发生和发展都起着重要的促进作用。本项目设计的目标分子将Trx-1抑制剂和端粒酶抑制剂的共能基团巧妙的设计在一起,获得了同时具有Trx-1抑制和端粒酶抑制作用的全新小分子化合物YLS004,体内外实验结果都表明其对结直肠癌和恶性黑色素瘤有非常显著的抑制作用,且毒性低。大鼠体内的药代动力学研究数据表明,大鼠静脉注射YLS004后其在大鼠体内能生成代谢物6-thio-dG,请见工作基础6。因此,YLS004的体内药代动力学过程和药效作用都符合我们对该类分子设计的思路,如下图所示。相关研究成果已经申请国家发明专利(专利名称:6-双硫取代-2’-脱氧鸟苷类化合物及其制备方法和应用,专利申请号201811003442.6)。

据2019年1月发布的癌症报告,我国2015年(统计报告晚3年)结直肠癌的发病人数是38.7万人,死亡人数为21.5万人。我国是全球胃肠道癌症的高发区,近几年结直肠癌的发病人数据估算已经远超该数值。目前临床上结直肠癌的常用治疗方案是5-FU联合奥沙利铂或伊立替康,但效果都不佳,特别是对中晚期病人。因此,结直肠癌新药的市场前景非常好。恶性黑色素瘤虽然临床上发病率较低,但转移性恶性黑色素瘤目前临床上没有好的治疗手段,一线用药达卡巴嗪疗效有限且毒副作用大。因此,开发疗效好、毒副作用低的结直肠癌和黑色素瘤治疗药物非常必要,同时也具有广阔的市场前景。
1.2.研发内容
本项目在前期部分成药性评价基础上,将按照新药临床前研究指导原则,对候选化合物YLS004开展系统的药学、药效学、安全性评价、药代动力学研究,内容如下:
(1)药学研究:利用正交试验优化YLS004的合成工艺,确定工艺参数,确定中试工艺研究;开展质量研究,制定药物质量标准,并开展化合物的稳定性研究;完善YLS004的制剂处方、工艺优化、确定制剂生产工艺参数,进行制剂质量标准研究。
(2)作用机制和药效学研究:采用重组酶、稳定高表达细胞系、基因Knockdown和化学抑制剂等技术开展Trx-1和端粒酶双靶点抑制研究,进一步明确作用机制;按照抗肿瘤新药药效学研究规范,以5-FU/奥沙利铂和达卡巴嗪等为阳性药物,以结直肠癌细胞和黑色素瘤细胞(包括耐药株)体外模型以及荷瘤鼠模型和PDX模型,系统研究YLS004对结直肠癌和黑色素瘤的抑瘤效果,获得剂量-药效关系。
(3)药代动力学研究:考察YLS004的代谢稳定性以及在动物体内的吸收、分布、代谢和排泄等PK特性,考察其对细胞色素P450酶和药物转运体的影响从而评估可能的药物相互作用。
(4)一般药理学研究:考察在有效剂量下,YLS004对动物的神经系统、心血管系统、呼吸系统等的作用。
(5)安全性评价研究:完成急性毒性实验(大鼠和犬)、长期毒性试验(6,12个月,大鼠和犬)、溶血性、局部刺激性、过敏性试验等,评价YLS004可能的毒性;进行毒代动力学研究。
2.关键技术:
(1)目标化合物的高效合成技术——针对二硫键基团发展YLS004的绿色高效合成技术,简化合成工艺,使用廉价、满足新药申报要求的原料,提高合成总收率、减少污染排放,满足新药申报要求。
(2)硫氧还蛋白和端粒酶抑制机制和药效评价技术——构建重组酶、稳定高表达细胞系、基因Knockdown等模型研究YLS004对硫氧还蛋白和端粒酶双靶点抑制的协同作用机制,评价量效关系。
- 3. 主要创新点
(1)该项目通过巧妙的分子设计,使得Trx-1和端粒酶两个热门抗癌药效靶点在一个分子上同时实现抑制,在细胞和动物模型上获得了对结直肠癌、恶性黑色素瘤等肿瘤效果显著的目标化合物YLS004,其药效明显优于而毒性明显低于在研的Trx-1抑制剂、6-thio-dG以及临床一线用药。
(2)YLS004具有全新分子结构,已申报国家发明专利(6-双硫取代-2’-脱氧鸟苷类化合物及其制备方法和应用,专利申请号201811003442.6)和PCT(美国)专利,具有完全自主知识产权。
据最新发布的癌症报告,我国(统计报告晚 3 年)结直肠癌的发病人数是大于 40 万人/年,死亡人数为超过 21.5 万人/年。我国是全球胃肠道癌症的高发区,近几年结直肠癌的发病人数据估算已经远超该数值。目前临床上结直肠癌的常用治疗方案是 5-FU 联合奥沙利铂或伊立替康,但效果都不佳,特别是对中晚期病人。因此,结直肠癌新药的市场前景非常好。恶性黑色素瘤虽然临床上发病率较低,但转移性恶性黑色素瘤目前临床上没有好的治疗手段,一线用药达卡巴嗪疗效有限且毒副作用大。血液系统癌症特别是晚期耐药性血液系统癌症,临床上还没有好的治疗方案。因此,开发疗效好、毒副作用低的结直肠癌、黑色素瘤和血液系统肿瘤治疗药物非常必要,同时也具有广阔的市场前景。

扫码关注,查看更多科技成果